Multi-Objective Generative Design of mRNA Therapeutics
mRNAutilus generates full-length mRNAs optimized for half-life, translation efficiency, and protein abundance, unifying codon optimization and de novo UTR design in a single generative process.
mRNAutilus generates full-length mRNAs optimized for half-life, translation efficiency, and protein abundance, unifying codon optimization and de novo UTR design in a single generative process.
Messenger RNA therapeutics have transformed medicine[1]. The COVID-19 pandemic proved that mRNA vaccines can be developed, manufactured, and distributed in mere months[2]. But beneath this success lies a persistent challenge: mRNAs are inherently unstable, and designing one that is simultaneously stable, efficiently translated, and highly expressed remains a multi-objective optimization problem that current methods struggle to address systematically.
An mRNA molecule is more than its coding sequence. The 5' untranslated region (UTR) initiates translation, the coding sequence (CDS) determines the protein product through codon choice, and the 3'UTR governs stability and localization. Critically, these regions interact: inter-region base-pairing, secondary structures at the 5'UTR-CDS junction, and 3'UTR structural elements all influence the overall fitness of an mRNA. Yet most existing design methods optimize each component in isolation.
Traditional codon optimization considers codons independently, substituting them with synonymous alternatives based on species-specific usage tables. More recent approaches use separate language models for each mRNA component — one for the CDS, one for the 5'UTR, one for the 3'UTR — and combine them post hoc. This component-by-component strategy requires lab-in-the-loop screening and misses the cross-region dependencies that determine overall function. Additionally, mRNA expression is highly dependent on the gene in consideration and differ markedly between tissues due to variations in intracellular machinery, metabolism, and innate immune activity.
Can we generate a complete, functional mRNA transcript — UTRs and optimized coding sequence together — in a single generative process, guided simultaneously toward multiple therapeutic properties?
mRNAutilus is the first framework to do exactly this, generating full-length mRNA transcripts with Pareto-optimal therapeutic properties through simultaneous codon optimization and de novo UTR design.
mRNAutilus — mRNA generation via Unrolled Trajectories and Informed Latent UpdateS — is a 150M-parameter masked discrete diffusion model[3,4] trained on 14.2 million full-length mRNA sequences from 342 vertebrate species[5]. It uses a hybrid tokenization scheme: codons in the CDS are tokenized as 3-mers, while UTR nucleotides are encoded individually, preserving the natural granularity of each region within a unified model.
The model starts from a fully masked sequence and progressively unmasks tokens, capturing bidirectional dependencies across the entire transcript. During conditional generation, a template protein coding sequence can be partially masked, allowing simultaneous codon optimization and de novo UTR generation. Importantly, biological constraints are enforced at every step: only synonymous codons (encoding the same amino acid) are permitted during CDS optimization, guaranteeing the designed mRNA produces the intended amino acid sequence.
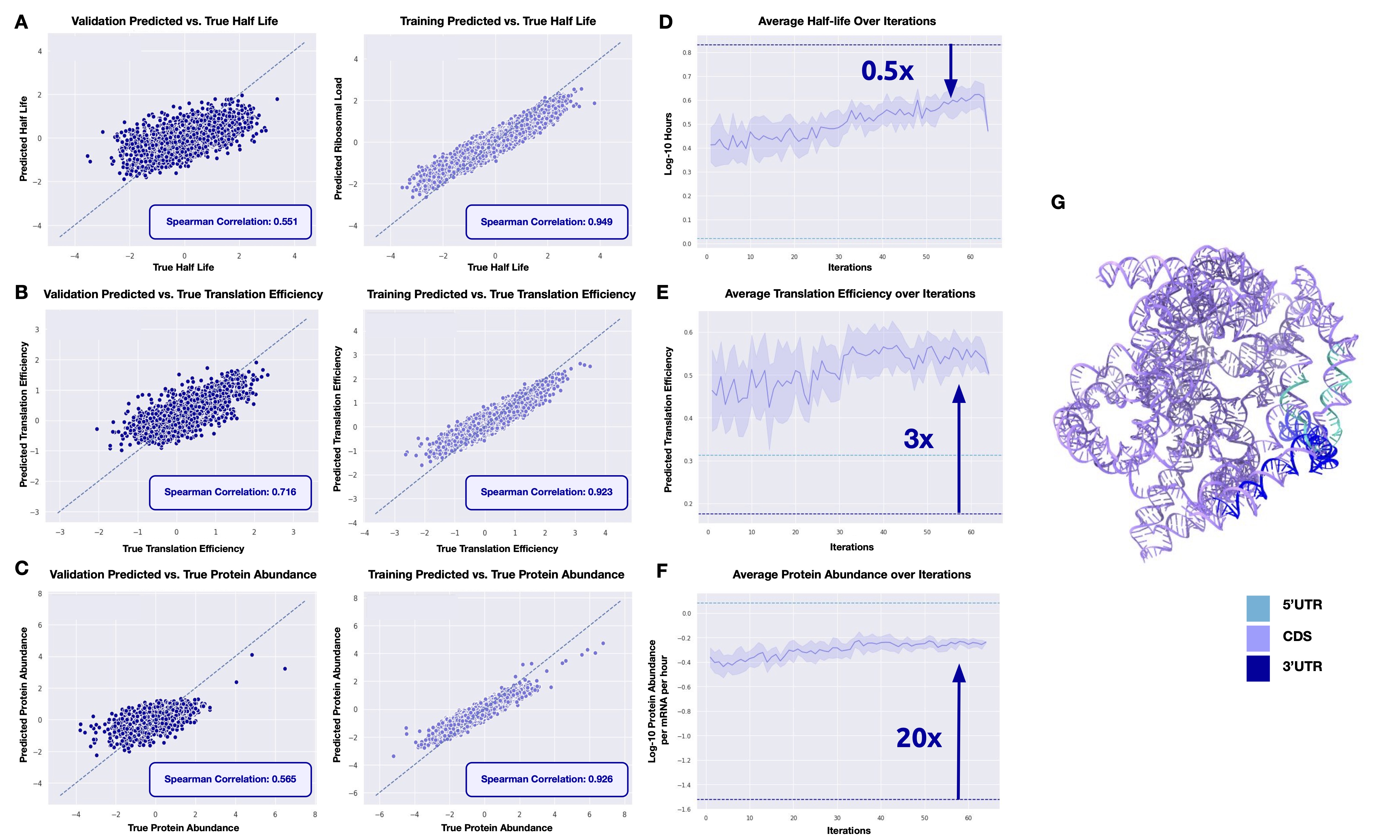
mRNAutilus embeddings serve two roles: they drive generation and enable lightweight property prediction. We train XGBoost regressors on mRNAutilus embeddings to predict three key mRNA properties from sequence alone, including half-life, translation efficiency, and protein abundance datasets used for guidance[9,10,11].
Despite their simplicity, these regressors are competitive with models up to 45× larger. On half-life and protein abundance, mRNAutilus (150M parameters) outperforms Helix-mRNA (6M), HyenaDNA (7M), and RiNALMo (150M), and matches the 7B-parameter Evo-2 model. These lightweight predictors serve as the reward functions that guide MCTG during generation — no gradient computation required.
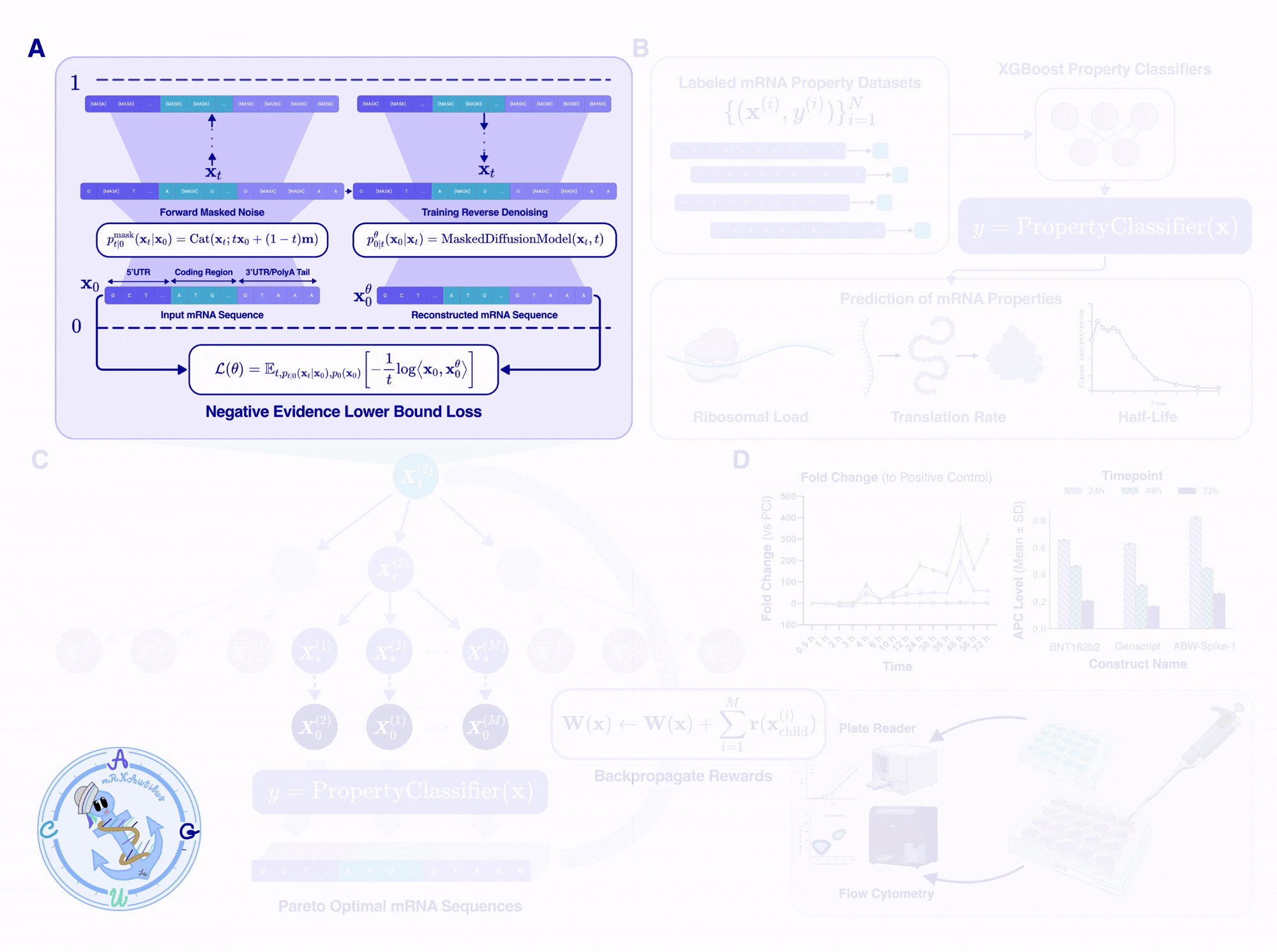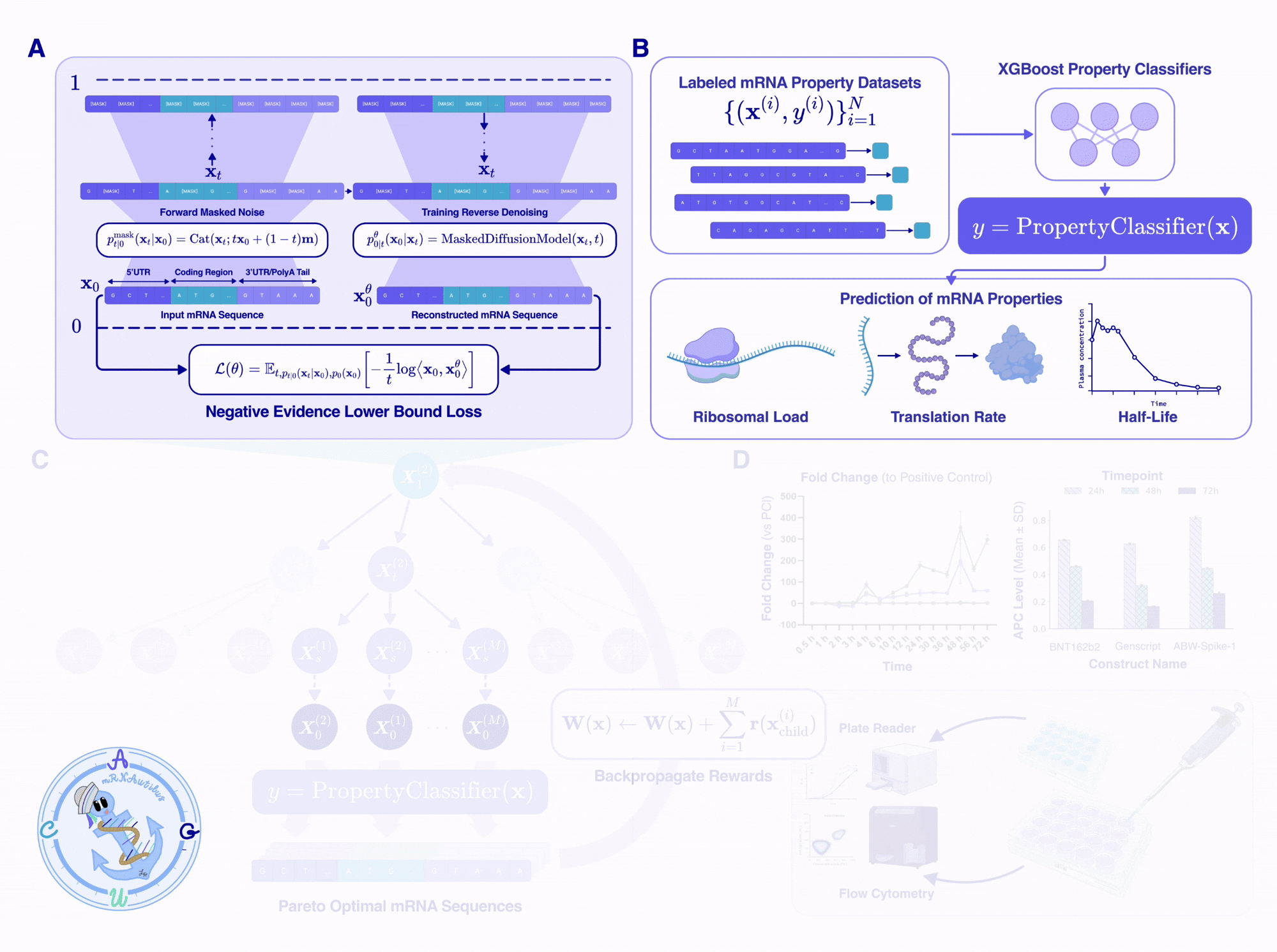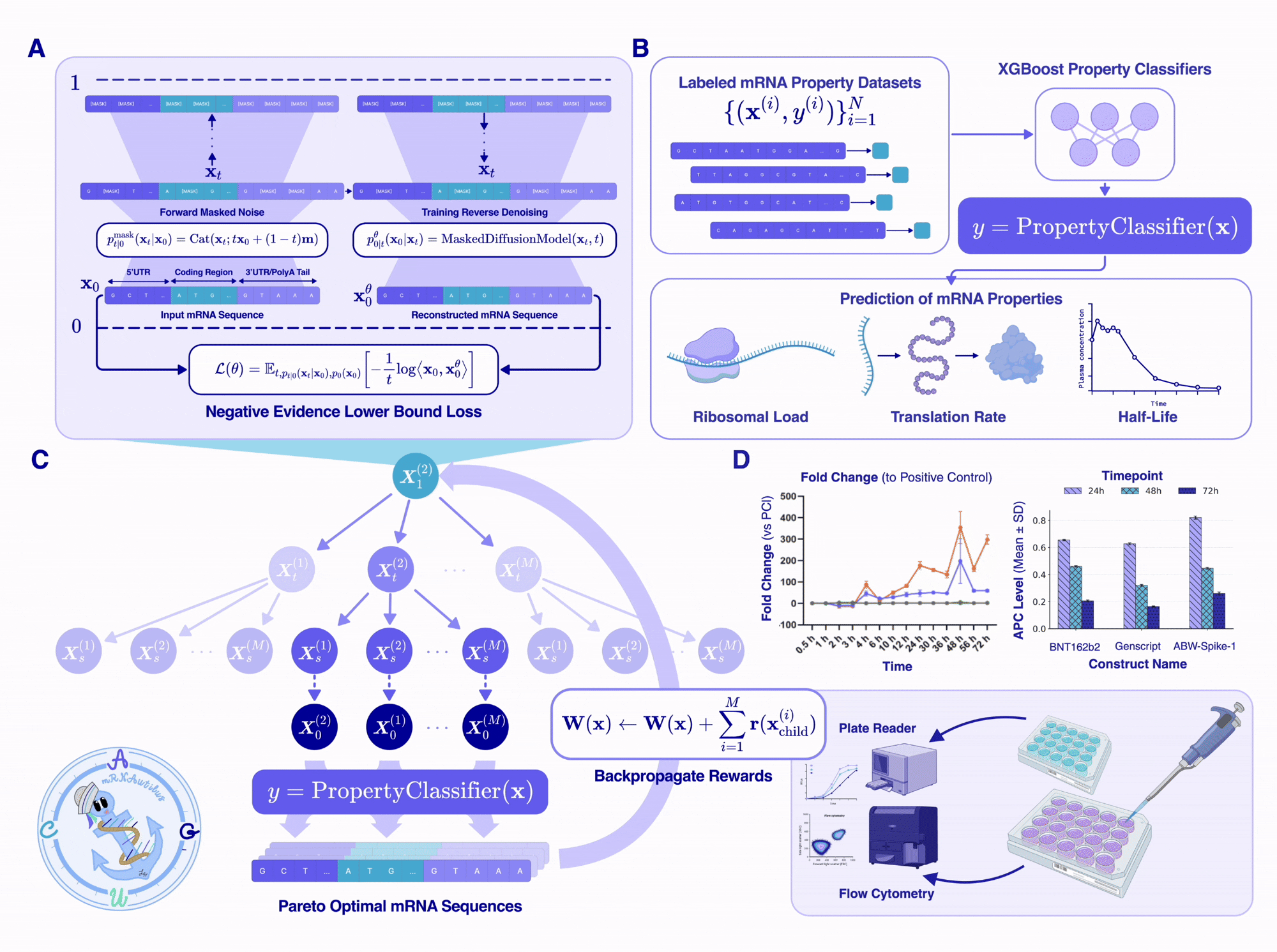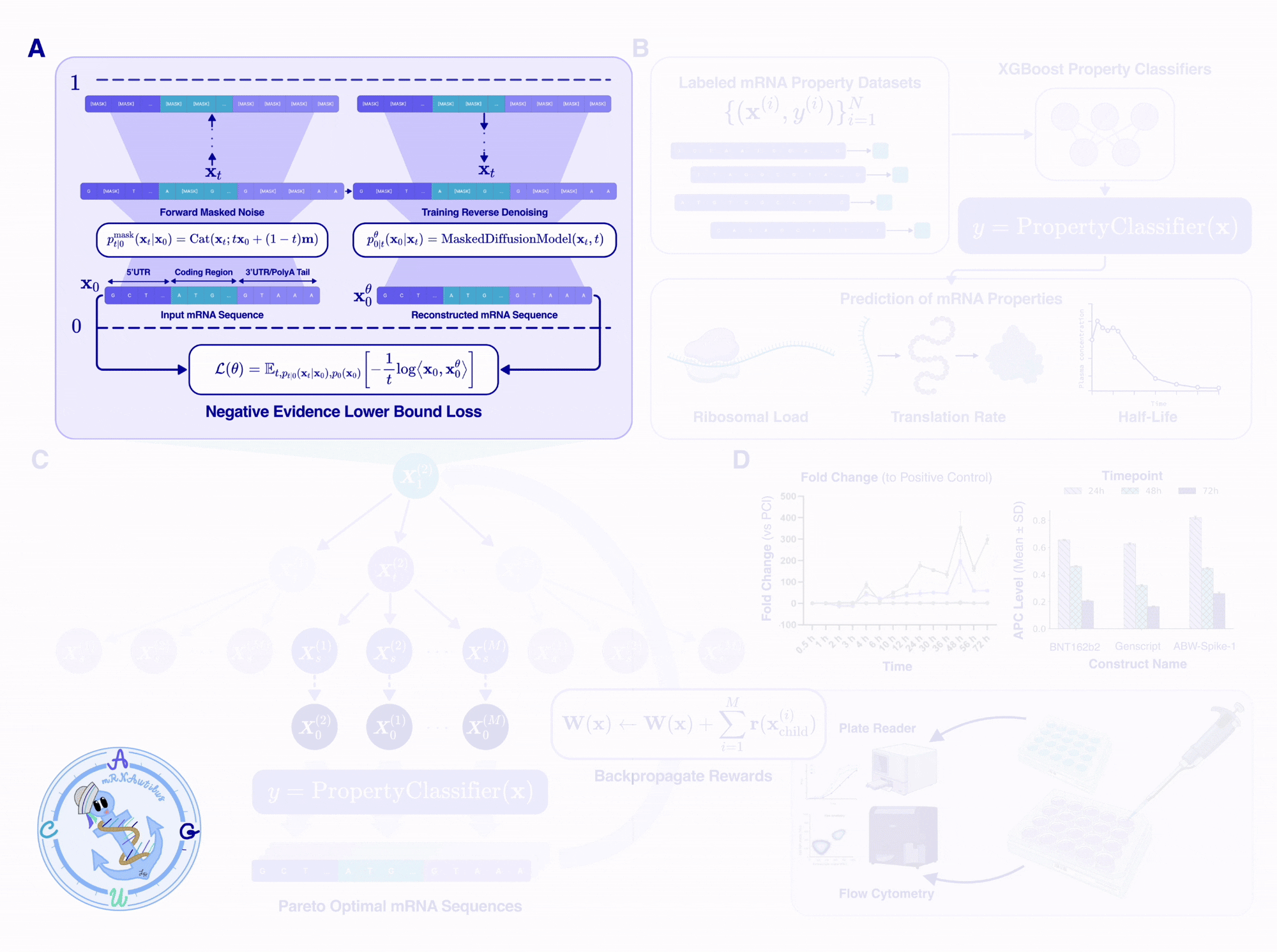
Designing a therapeutic mRNA requires optimizing multiple competing properties simultaneously — half-life, translation efficiency, and protein abundance each pull the sequence in different directions. Rather than optimizing a single scalar objective, mRNAutilus frames mRNA design as a multi-objective optimization problem and searches for the set of Pareto-optimal solutions: sequences where no property can be improved without degrading another.
To navigate this trade-off landscape, we leverage Monte Carlo Tree Guidance (MCTG)[6], a gradient-free algorithm that steers the masked diffusion model's denoising process toward Pareto-optimal mRNA sequences. MCTG treats each step of the unmasking trajectory as a node in a search tree and uses Monte Carlo rollouts to evaluate which unmasking decisions lead to the best multi-property outcomes.
The MCTG algorithm operates in four steps, which iteratively optimize multi-objective rewards:
At every expansion step, MCTG enforces hard biological constraints to guarantee valid mRNA output. Codon tokens cannot appear in UTR positions, and for any masked codon, only synonymous codons encoding the same amino acid as the template are permitted. This ensures the designed mRNA always encodes the intended amino acid sequence.
The result is a set of non-dominated mRNA sequences spanning the Pareto front across half-life, translation efficiency, and protein abundance. Researchers can then select from this front based on their application-specific priorities — for instance, favoring durability for a vaccine or peak expression for a reporter assay.
Unlike gradient-based guidance that requires differentiable reward functions, MCTG works with any black-box predictor — including the lightweight XGBoost regressors trained on mRNAutilus embeddings. This makes multi-objective guidance practical even in low-data settings where training a differentiable surrogate would be unreliable.
Before applying property guidance, we verified that mRNAutilus generates biologically plausible mRNA sequences. Libraries of unconditionally generated mRNAs were compared against vertebrate mRNAs from the Ensembl training corpus across multiple biophysical metrics.
Generated sequences exhibit higher GC content (a hallmark of efficient translation), lower minimum free energy (indicating greater thermodynamic stability), and natural Kozak consensus frequencies. Critically, the generated libraries maintain sequence diversity comparable to natural mRNAs, with nearly identical Shannon entropy distributions — ruling out degenerate repetitive sampling. The model learns to produce diverse, stable, translation-ready sequences without any explicit optimization.
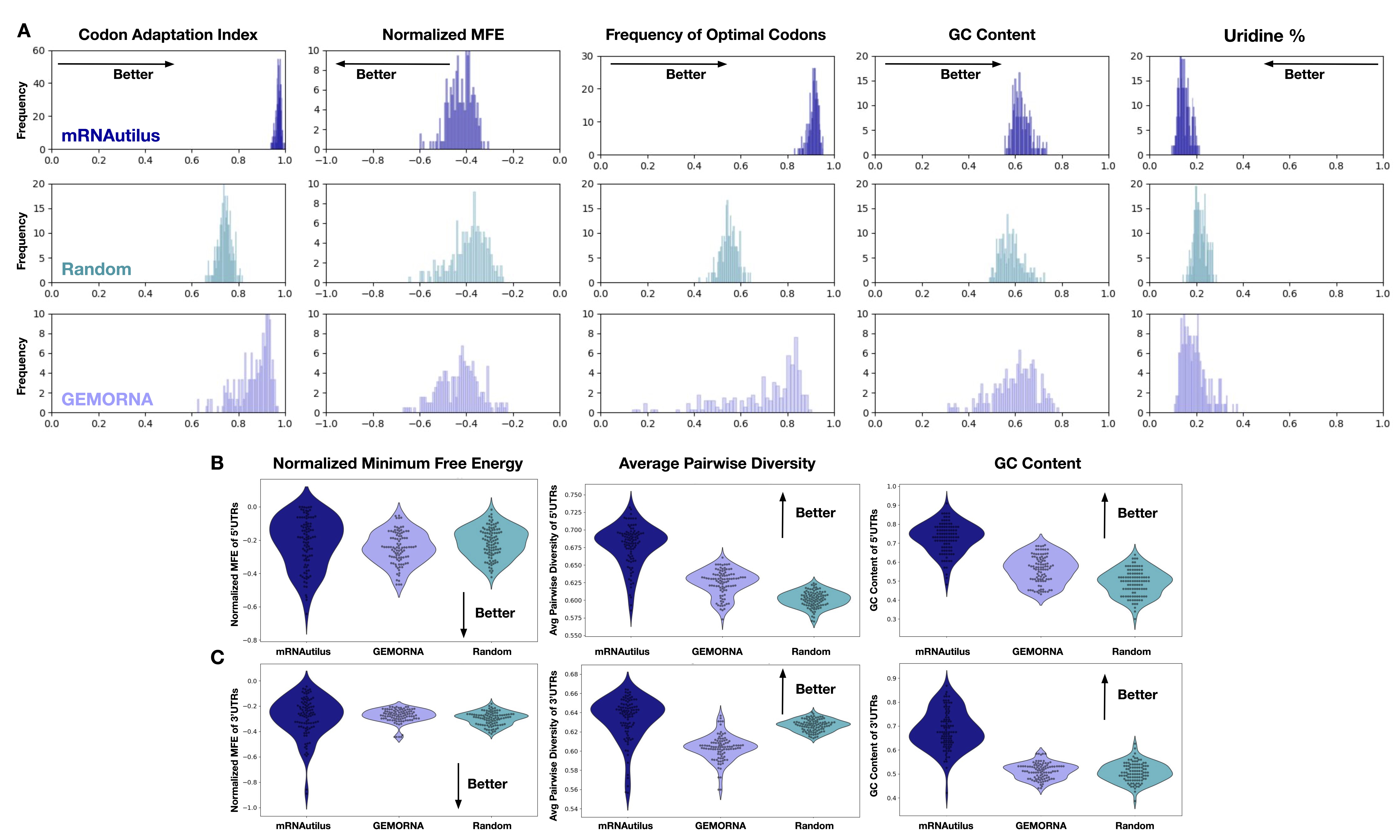
We compared mRNAutilus-designed full-length luciferase mRNAs against sequences produced by GEMORNA[7] (an autoregressive component-by-component approach) and random sampling. Across codon adaptation index, minimum free energy, optimal codon frequency, and uridine percentage, mRNAutilus-designed CDS sequences achieved consistently improved metrics. Generated UTRs showed greater thermodynamic stability and diversity than both alternatives.
The true test of any generative model is the wet lab. mRNAutilus-designed mRNAs were synthesized and tested in vitro across three targets with distinct biological and therapeutic relevance — P. pyralis luciferase, SARS-CoV-2 spike glycoprotein, and PEMax (a prime-editor payload). All designs were generated zero-shot — without any iterative laboratory optimization — drawn from libraries jointly optimized for half-life and translation efficiency, and benchmarked against human alpha-globin (HAB) UTR composition as an in silico baseline. For each target, the top five designs by predicted fold-change over the HAB baseline were synthesized by in vitro transcription and evaluated in cell-based expression assays.
Zero-shot mRNAutilus designs produced luminescence signals comparable to or significantly greater than wild-type control in HEK293T cells, with designed sequences exhibiting approximately 400-fold higher expression than wild-type P. pyralis luciferase at 48h post-transfection. Across three additional cell lines (Jurkat, A549, HepG2), the same mRNAutilus designs consistently outperformed both zero-shot GEMORNA-designed luciferase mRNA (GMR-FL) and the composition combining GenScript F-Luc CDS with human alpha-globin UTRs.
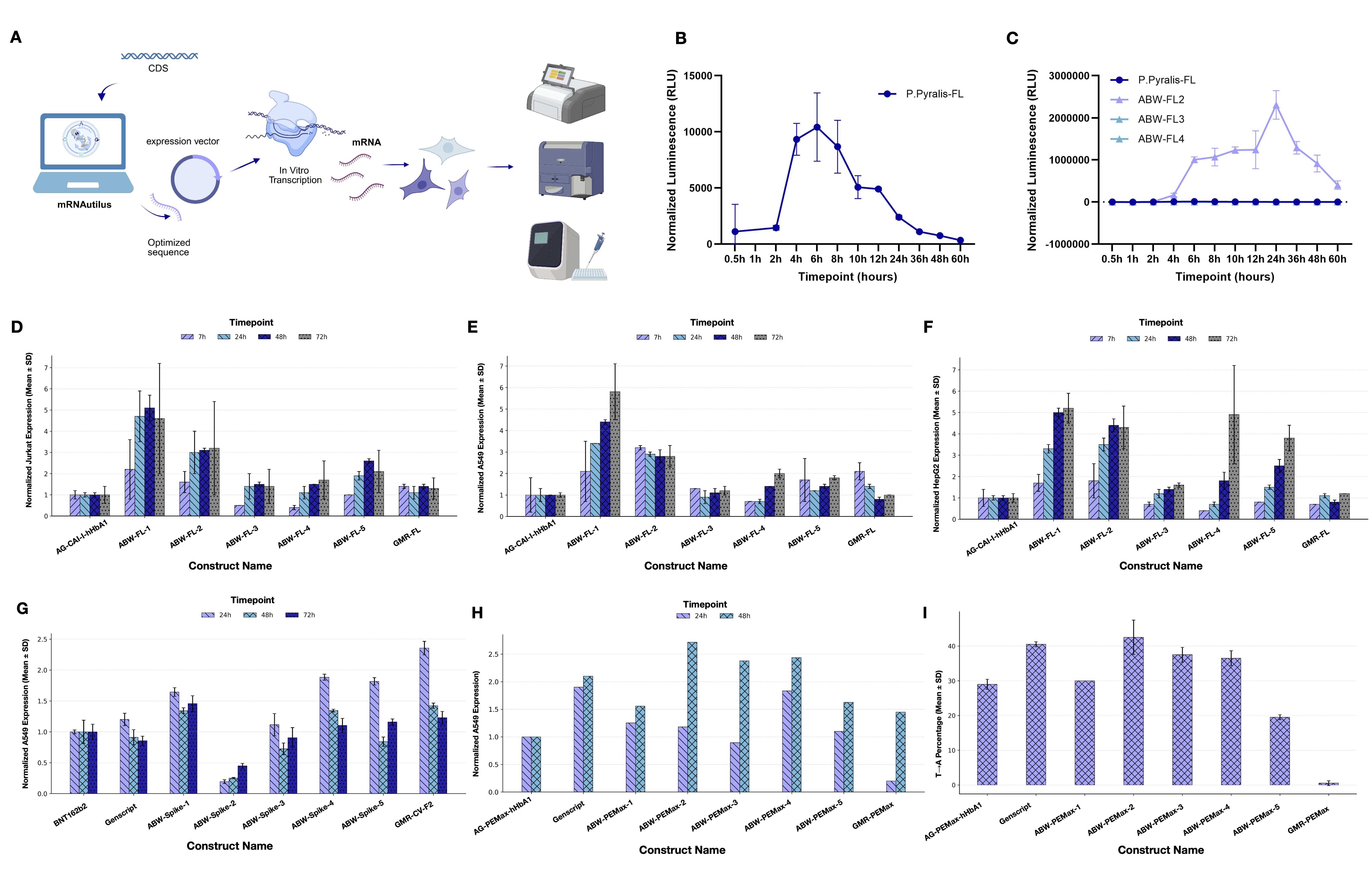
Three zero-shot mRNAutilus designs (ABW-Spike-1, ABW-Spike-2, ABW-Spike-3) expressed above both BNT162b2 (Pfizer/BioNTech) and the commercial GenScript Spike v2 mRNA in A549 cells. Across all measured time points, these designs matched the expression profile of the lab-in-the-loop multi-shot GEMORNA sequence (GMR-CV-F2), with ABW-Spike-1 additionally exhibiting improved intracellular stability.
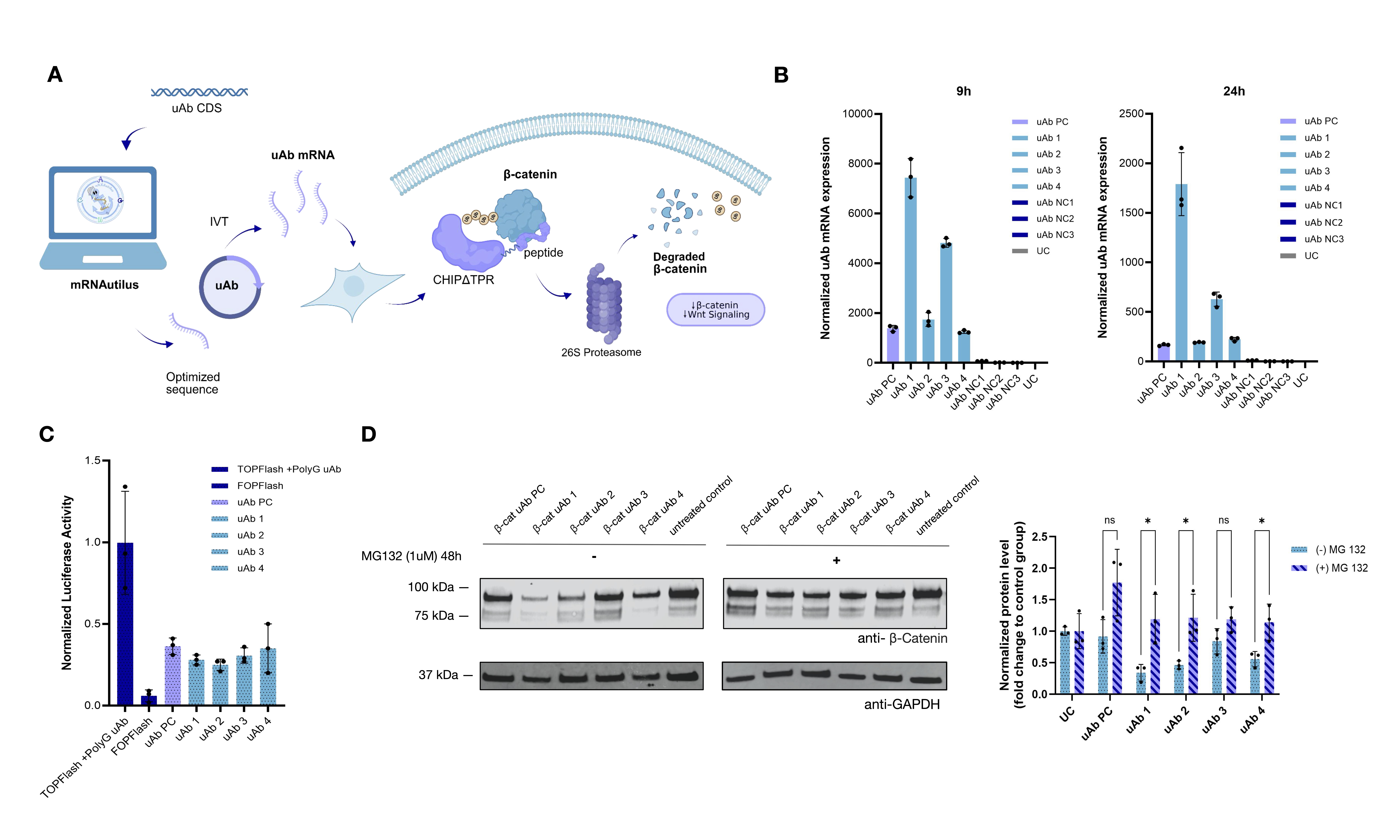
mRNAutilus-designed constructs for prime editing (PEMax) similarly outperformed baselines, highlighted by more durable expression over commercial mRNAs and improved gene editing efficiency. Beyond reporters, antigens, and enzymes, we further demonstrated generality to functionally demanding intracellular payloads using ubiquibodies (uAbs) — synthetic E3 ligases where the substrate-binding domain of CHIP is replaced with peptide binders[12], enabling programmable degradation of disease-relevant targets. mRNAutilus-designed uAb mRNAs targeting β-catenin showed enhanced expression and functional efficacy, establishing the framework as broadly applicable across diverse therapeutic modalities.
mRNAutilus-designed uAb mRNAs targeting β-catenin showed markedly higher transcript abundance than the human α-globin-based positive control at both 9 h and 24 h post-transfection in DLD-1 cells. Optimized constructs fully retained downstream degrader function in the TOPFlash Wnt/β-catenin reporter assay, and immunoblot analysis confirmed significant proteasome-dependent reductions in endogenous β-catenin for uAb1, uAb2, and uAb4 (reversed by MG132 co-treatment) — establishing mRNAutilus as broadly applicable to programmable intracellular therapeutic modalities.
We benchmarked mRNAutilus embeddings for property prediction against leading nucleic acid language models, training identical XGBoost regressors on each model's embeddings. The key result from the manuscript is not a single translation-efficiency score, but the full tradeoff: mRNAutilus delivers the strongest half-life prediction and is effectively tied for best protein abundance while using far fewer parameters than Evo-2.
mRNAutilus is the top model on half-life prediction (R² = 0.551), exceeding Evo-2 and Helix-mRNA despite being far smaller than Evo-2. On protein abundance, it is essentially tied for best overall, trailing Evo-2 by just 0.001 (0.565 vs. 0.566). Evo-2 and Helix-mRNA embeddings are demonstrably better for predicting translation efficiency, yet mRNAutilus remains competitive while offering the strongest overall balance across all three tasks.
mRNAutilus establishes a new paradigm for mRNA design: a single generative process that jointly optimizes codon usage, UTR sequences, and their cross-region interactions toward multiple therapeutic properties. Unlike prior methods that design components in isolation or require iterative laboratory screening, mRNAutilus generates complete, ready-to-synthesize transcripts zero-shot.
Future directions include incorporating additional property regressors for immunogenicity and tissue-specific expression, integrating manufacturing constraints directly into the guidance procedure, exploring non-uniform reward weighting, and coupling with high-throughput screening for design-build-test-learn cycles. The framework is gradient-free and practical even in low-data settings, opening the door to rapid mRNA therapeutic design across diverse biological applications.
mRNAutilus is a significant step toward a unified framework for controllable, multi-objective-guided, full-length mRNA therapeutic design.